When you pick up a bottle of generic lisinopril at your local pharmacy, you probably don’t think about the years of paperwork, lab tests, and negotiations that got it there. But behind every cheap, effective generic drug is a complex journey-from a regulatory application filed with the FDA to the shelf next to the brand-name version. This is the real story of how generic drugs make it from approval to your medicine cabinet.
What Is an ANDA, and Why Does It Matter?
An Abbreviated New Drug Application (ANDA) is the official form generic drug makers submit to the U.S. Food and Drug Administration (FDA) to get approval to sell a copy of a brand-name drug. It’s called ‘abbreviated’ because it doesn’t require repeating the expensive, years-long clinical trials that the original drug went through. Instead, the generic company must prove their version is bioequivalent-meaning it delivers the same amount of active ingredient into the bloodstream at the same rate as the brand-name drug.
The ANDA pathway was created by the Hatch-Waxman Act in 1984. Its goal? Lower drug prices without sacrificing safety. Today, nearly 90% of all prescriptions filled in the U.S. are for generics. And they save patients and the healthcare system an estimated $313 billion every year.
But getting that approval isn’t easy. A complete ANDA includes detailed information on the drug’s chemistry, how it’s made, how stable it is, and proof of bioequivalence. It also needs identical labeling to the brand-name drug-except for the name and manufacturer. Even small mistakes, like a typo in the dosage instructions or an incomplete manufacturing plan, can trigger a ‘Complete Response Letter’ from the FDA. About 40% of initial ANDA submissions get rejected on the first try.
The Approval Process: From Submission to Green Light
Submitting an ANDA isn’t just filling out a form. It’s a high-stakes, highly technical process. All applications must be filed electronically through the FDA’s Electronic Submissions Gateway. The agency reviews the data under the Generic Drug User Fee Amendments (GDUFA), a system that sets clear timelines and fees to keep the process moving.
For a standard generic drug, the FDA aims to complete its review within 30 months. But if the drug is a first generic version of a brand-name product-or if it’s needed to fix a shortage-the FDA can fast-track it. In 2022 alone, the agency approved 112 first generics, covering drugs with $39 billion in annual U.S. sales.
Complex generics-like inhalers, patches, or injectables-take longer. They’re harder to copy because their delivery method matters as much as the chemical. These products have a lower approval rate: about 65%, compared to 85% for simple pills. That’s why many companies hold pre-ANDA meetings with the FDA. These early discussions help avoid costly mistakes. Companies that use them see 30% fewer major issues during review.
There’s also the patent game. If a generic company believes a brand-name patent is invalid or doesn’t apply, they can file a ‘Paragraph IV certification.’ This challenges the patent and can trigger a 30-month legal delay. But if they win, they get 180 days of exclusive rights to sell the first generic version. That’s why, when the patent for Eliquis (apixaban) was set to expire in 2022, six different companies rushed to file ANDAs at the same time.

Approval Doesn’t Mean Availability
Many assume that once the FDA says ‘yes,’ the drug hits store shelves. But that’s only the halfway point. In fact, 78% of generic manufacturers say the real challenge begins after approval.
First, production must scale up. A lab batch of 10,000 pills is one thing. A commercial run of 10 million is another. Factories need to adjust equipment, train staff, and prove they can consistently meet quality standards. This transition takes 60 to 120 days.
Then comes the biggest hurdle: getting paid for it. Generic drugs don’t sell directly to pharmacies. They go through pharmacy benefit managers (PBMs)-companies like Express Scripts, OptumRx, and CVS Health that negotiate prices with drug makers on behalf of insurers and employers.
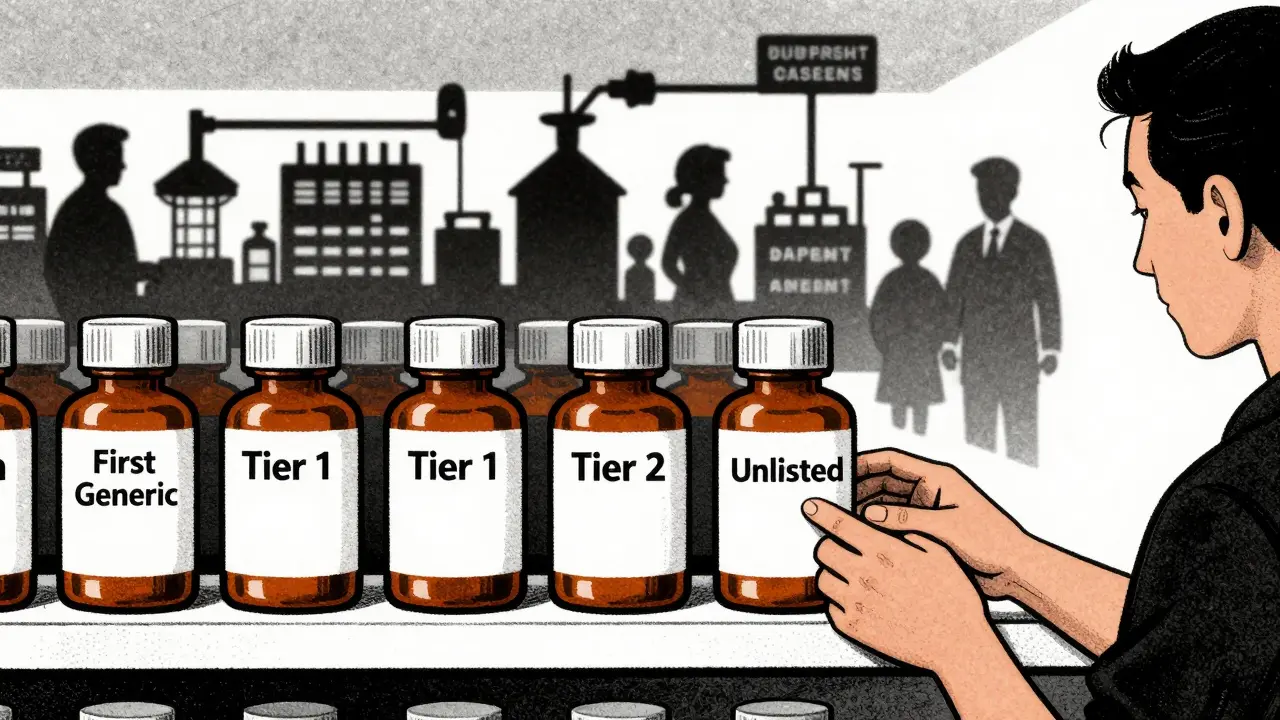
These PBMs decide which generics go on which ‘tier’ of a formulary. Tier 1 is the cheapest, preferred option. Tier 2 costs more. If your generic isn’t on Tier 1, most patients won’t choose it. To get there, manufacturers often have to offer discounts of 20-30% deeper than they planned. One sourcing manager on Reddit put it bluntly: ‘If you’re not on the top three tiers, your product might as well not exist.’
Getting It to the Pharmacy
Once pricing is locked in, the drug moves into distribution. Most generics use the big three wholesalers: AmerisourceBergen, McKesson, and Cardinal Health. These companies handle logistics for 90% of U.S. pharmacies. But adding a new product to their system isn’t instant. It takes 15 to 30 days to update inventory codes, shipping labels, and tracking systems.
Then, the pharmacy itself has to receive the shipment, scan it into their system, and train staff to answer patient questions. For most drugs, this final step takes less than two weeks. But for high-volume medications-like blood pressure pills or statins-the whole process from FDA approval to first sale averages just 87 days.
For complex drugs-like asthma inhalers or topical creams-it can take over 145 days. Why? More steps. More testing. More negotiations. And often, fewer competitors, which means less pressure to move quickly.
Why This System Works-and Why It’s Under Pressure
The ANDA system has delivered massive savings. Since 2013, generic drugs have saved the U.S. healthcare system over $1.67 trillion. In 2022, Americans filled 6.3 billion generic prescriptions. The market is now worth $124.7 billion.
But things are changing. Generic drug prices have dropped an average of 4.7% every year since 2015. That’s good for patients-but bad for manufacturers. Profit margins are shrinking. Some companies have stopped making certain low-margin generics, leading to shortages.
The FDA is responding. In 2023, they launched the Drug Competition Action Plan to prevent anti-competitive practices. They’re also pushing for better quality control, especially for drugs made overseas. A new rule, effective January 2024, requires standardized electronic submissions, which should speed things up-but only if companies can adapt.
Some experts predict artificial intelligence will soon help design bioequivalence studies and predict manufacturing issues, cutting development time by 25-30%. But regulators are cautious. AI can’t replace human review-not yet.
What’s Next for Generic Drugs?
By 2028, generics are expected to make up 93% of all prescriptions. That’s up from 90% today. But the path to get there is getting harder. More complex drugs are coming off patent-biologics, inhalers, injectables. These aren’t easy to copy. The FDA is creating new guidelines just to handle them.
Meanwhile, manufacturers are turning to the 505(b)(2) pathway for some products-this lets them tweak existing drugs slightly to get faster approval. It’s not a true ANDA, but it’s becoming a popular workaround.
One thing’s clear: the system works, but it’s fragile. A single factory shutdown overseas, a patent lawsuit, or a PBM refusing to list a drug can delay access for thousands. The goal is still the same: safe, affordable medicine. But the journey from ANDA to shelf is longer, tougher, and more competitive than most people realize.
How long does it take for a generic drug to reach the pharmacy after FDA approval?
On average, it takes about 112 days from FDA approval to the first retail sale. But this varies widely. Simple pills like generic statins or blood pressure meds can be on shelves in 80-90 days. Complex products like inhalers or patches often take 140 days or more due to longer manufacturing and distribution steps.
Why are generic drugs so much cheaper than brand-name drugs?
Generic drugs cost 80-85% less because they don’t repeat the expensive clinical trials the original drug went through. Instead, they prove bioequivalence using smaller, targeted studies. Development costs average $2-5 million for a generic, compared to $2.6 billion for a new brand-name drug. Plus, competition among multiple generic makers drives prices down further.
Can a generic drug be different from the brand-name version?
The active ingredient must be identical, and the drug must deliver the same amount into the bloodstream at the same rate. But generics can have different inactive ingredients-like fillers, dyes, or coatings. These don’t affect how the drug works, but they might cause rare allergies or affect how the pill looks or tastes. That’s why some patients report feeling different on a generic-usually due to psychological factors or minor variations in absorption.
What is a ‘first generic’ and why does it matter?
A ‘first generic’ is the very first generic version of a brand-name drug approved after its patent expires. The first company to file a complete ANDA with a Paragraph IV certification gets 180 days of market exclusivity-no other generics can enter during that time. This period is extremely valuable because it allows the first maker to capture the majority of sales before competition kicks in. In 2022, 112 first generics were approved, covering drugs worth $39 billion in annual sales.
Do all pharmacies stock every generic drug?
No. Pharmacies stock generics based on what their pharmacy benefit manager (PBM) allows and what’s most cost-effective. If a generic isn’t on the preferred tier of a PBM’s formulary, many pharmacies won’t order it-even if it’s FDA-approved. Patients may need to ask for a specific generic or switch to a different pharmacy to get the one they want.
11 Comments
Ryan HutchisonJanuary 18, 2026 AT 04:07
Let me break this down real simple - the FDA’s ANDA process is a joke. You spend millions on a drug, then some overseas factory with half the equipment and zero QA gets approved because they paid the right people. And we wonder why we have shortages? It’s not the system working - it’s the system being gamed by Chinese and Indian labs that don’t even follow GMP. We’re outsourcing our healthcare to countries that don’t even have clean water in some regions. Wake up, America.
And don’t get me started on PBMs. Those middlemen are the real drug price killers. They take 30% off the top and still act like they’re doing us a favor. The whole system is rigged.
Meanwhile, the FDA sits on their hands waiting for AI to fix everything. Newsflash - AI can’t fix corruption. Only real oversight can.
And yeah, I’ve seen generics that made me dizzy. Not placebo. Real difference. Because the inactive ingredients? Totally different. They’re not just ‘fillers’ - they’re chemical wildcards. FDA says it’s fine. I say I’m not a lab rat.
Stop pretending this is about ‘affordability.’ It’s about profit for conglomerates and political favors. We need a national generic drug manufacturing initiative. Not more paperwork. More factories. On American soil.
And if you think a 140-day delay is normal, you haven’t been paying attention. That’s a national security risk. What if it’s a heart med? A kid’s antibiotic? We’re gambling with lives because we’re too lazy to build our own supply chain.
And don’t even start with ‘but it’s cheaper!’ Cheap doesn’t mean safe. Cheap means you’re getting a product made in a factory where the QA guy got fired last week because he asked too many questions.
Stop glorifying the ANDA system. It’s a Band-Aid on a gunshot wound.
Melodie LesesneJanuary 19, 2026 AT 01:09
Wow, this was actually really eye-opening! I had no idea how much goes into getting a generic on the shelf. I always just thought, ‘oh it’s the same pill, cheaper’ - but the whole PBM tier system? That’s wild. No wonder I can’t find my usual generic sometimes.
Also, the part about first generics getting 180 days exclusivity? That’s kind of genius. It’s like a reward for being the first to take the risk. Makes sense, honestly.
Thanks for writing this - I’m gonna start asking my pharmacist more questions now. Maybe I’ll even look up which tier my meds are on. Who knew pharmacy was this complicated?
Corey SawchukJanuary 20, 2026 AT 06:04
Yeah the 87 day average sounds right for the simple stuff like lisinopril
but for inhalers or patches? Man that’s a whole other beast
my mom’s asthma inhaler took 6 months after approval to show up
and when it did it was like 30% more expensive than the brand even though it was supposed to be generic
guess the PBM didn’t like the new maker
so we just kept paying the brand price
not cool
Jody FahrenkrugJanuary 20, 2026 AT 18:25
I’ve been on the same generic blood pressure med for 8 years and never thought about any of this. Honestly, I just take it and forget. But now I’m kinda curious - do you think the inactive ingredients could be why I get headaches sometimes when I switch bottles?
My pharmacy swaps it out all the time and I’ve noticed I feel off for a few days after. I thought it was just stress, but now I wonder if it’s the dye or something.
Also, huge thanks for explaining the PBM thing. I thought pharmacies just picked what to stock. Turns out they’re basically told what to carry. That’s messed up.
Kasey SummererJanuary 21, 2026 AT 22:25
So let me get this straight - we spend $2.6B to make a brand drug, then some dude in Bangalore copies it for $5M and we call it ‘affordable’? 🤡
And the FDA approves it because… the math adds up? Cool cool cool.
Meanwhile, I’m paying $4 for my generic metformin but my insurance still charges me $20 copay because the PBM ‘negotiated’ a better deal with the brand.
Y’all realize we’re all just NPCs in a corporate simulation, right? 😎
At least the AI will take over soon. Maybe it’ll fix the system before we all die of preventable hypertension.
evelyn welldingJanuary 22, 2026 AT 20:22
This is so cool!! 🙌 I had no idea how much work went into making cheap meds possible. Like, I just thought generics were ‘copies’ but this is actual science + bureaucracy + negotiation magic! 💊
And the part about first generics getting 180 days exclusivity? That’s like a reward for being brave! So cool that someone’s risking it to make things cheaper for everyone.
Also, thanks for explaining why my pharmacy doesn’t have my favorite generic - I thought they were just being annoying. Now I know it’s the PBM. Ugh. But at least now I know what to ask for 😊
Keep sharing stuff like this - you’re making the world a little more understandable!
Bianca LeonhardtJanuary 24, 2026 AT 14:36
People who think generics are ‘just as good’ are either naive or paid by Big Pharma. The FDA’s bioequivalence standard is a joke. 80-125% range? That’s a 45% swing in absorption. You’re not getting the same drug - you’re getting a version that might work… sometimes.
And don’t get me started on the manufacturing. Most generics are made in India or China. Factories with mold on the walls and no real QA. The FDA inspects once every 5 years. That’s not oversight. That’s negligence.
And you think the PBM system is fair? Ha. They’re just middlemen who squeeze manufacturers until they go bankrupt. Then they jack up prices on the few left.
This system isn’t broken. It’s designed to fail. So we keep paying for brand names. And you’re all too dumb to see it.
Christina BilottiJanuary 24, 2026 AT 15:17
Oh wow, another ‘educational’ piece on how generics are ‘so cheap’ - how quaint. Let me guess, you also think ‘organic’ means ‘not sprayed with pesticides’ and ‘natural’ means ‘not made in a lab’? How endearing.
The FDA’s ‘bioequivalence’ is a statistical loophole. 80-125%? That’s not medicine. That’s gambling. You’re not getting the same drug - you’re getting a version that might work if you’re lucky and your liver is feeling generous.
And the ‘first generic’ exclusivity? That’s not innovation. That’s rent-seeking. The company that files first doesn’t invent anything - they just read the patent and race to the finish line like a dog chasing a car.
Meanwhile, real innovation - like new molecular entities - is being strangled by this system. Why spend $2.6B on a novel drug when you can copy one for $5M and get 180 days of monopoly? It’s not capitalism. It’s a Ponzi scheme disguised as healthcare.
And you think AI will fix this? Please. AI doesn’t fix greed. It just makes it faster.
Go back to your $4 metformin and stop pretending this is a success story.
brooke wrightJanuary 25, 2026 AT 13:52
I just switched from brand to generic and now I’m having weird dreams and my skin is itchy and I think my hair is falling out and I don’t know if it’s the generic or my cat or my ex or the new laundry detergent but I’m 37 and this is the first time I’ve ever felt like my body is betraying me and I’m scared and I don’t know who to call and I think the pharmacist said the generic was made in India but I don’t even know what that means and I just want to go back to the brand but my insurance won’t cover it and I’m crying in the parking lot of Walmart and I don’t know if anyone else feels this way or if I’m just crazy but I swear I can feel the difference and I don’t know if I’m being dramatic or if this is real but I just need someone to say it’s okay to be scared and I don’t want to be a burden but I don’t know what to do anymore
Nick ColeJanuary 27, 2026 AT 12:02
Just wanted to say thank you for writing this. I’ve worked in hospital pharmacy for 12 years and this is exactly what we see every day. The system is a mess - but not because of the people. It’s because of the structure.
My favorite part? The ‘first generic’ exclusivity. That’s the one thing that actually works. It’s the only incentive that pushes companies to take the risk. Without it, no one would bother.
And yeah, the PBM thing? Brutal. I’ve had patients cry because their med disappeared from the formulary. Not because it was unsafe. Just because the PBM didn’t like the price.
We’re not just dispensing pills. We’re navigating a maze of corporate contracts. And patients are the ones paying the price - literally and emotionally.
Keep pushing for transparency. We need more people who understand this.
Riya KatyalJanuary 29, 2026 AT 02:26
So the FDA approves a generic, then the PBM says ‘nope, not on our list’ and the whole thing dies? That’s not a market. That’s a dictatorship. And we call this ‘free enterprise’? LOL.
Meanwhile, in India, my cousin’s factory makes 10 million lisinopril pills a day and the boss doesn’t even know what bioequivalence means. He just checks the color and ships it.
But hey - it’s cheap. So we’re all winners. 🤷♀️
Also, why does the US still think it’s the only country that can make medicine? We import 80% of our antibiotics. We’re just a giant pharmacy with a flag.