
For those unfamiliar, Bioequivalence is the scientific demonstration that a generic product delivers the same amount of active substance at the same rate as the reference listed drug. In the world of BE testing, the goal is to show that the generic is therapeutically equivalent without having to run a massive, expensive clinical trial on thousands of patients. While this sounds straightforward, combination products introduce variables that can derail even the most experienced development teams.
The Fixed-Dose Combination (FDC) Headache
When you put two or more active pharmaceutical ingredients (APIs) into one pill, you've created a Fixed-Dose Combination (or FDC). The biggest risk here is chemical interference. One drug might change how the other dissolves, or they might compete for the same absorption transporters in the gut. If you're combining insoluble molecules, the risk of erratic absorption is huge.
Regulatory bodies aren't taking shortcuts here. To prove an FDC works, developers usually have to show BE against both the individual mono-products and the co-administered reference products. This often means using a three-way crossover protocol. Instead of a simple A-B test, you're managing more subjects-often 40 to 60 volunteers compared to the usual 24 for a single-entity drug-to ensure the statistical power is high enough to catch variability between the two drugs.
It's a high-stakes game. Data shows that FDC studies fail 25-30% more often than single-entity products. Why? Because a formulation that works for Drug A might inadvertently hinder the release of Drug B, leading to a failure in the 90% confidence interval for Cmax or AUC.
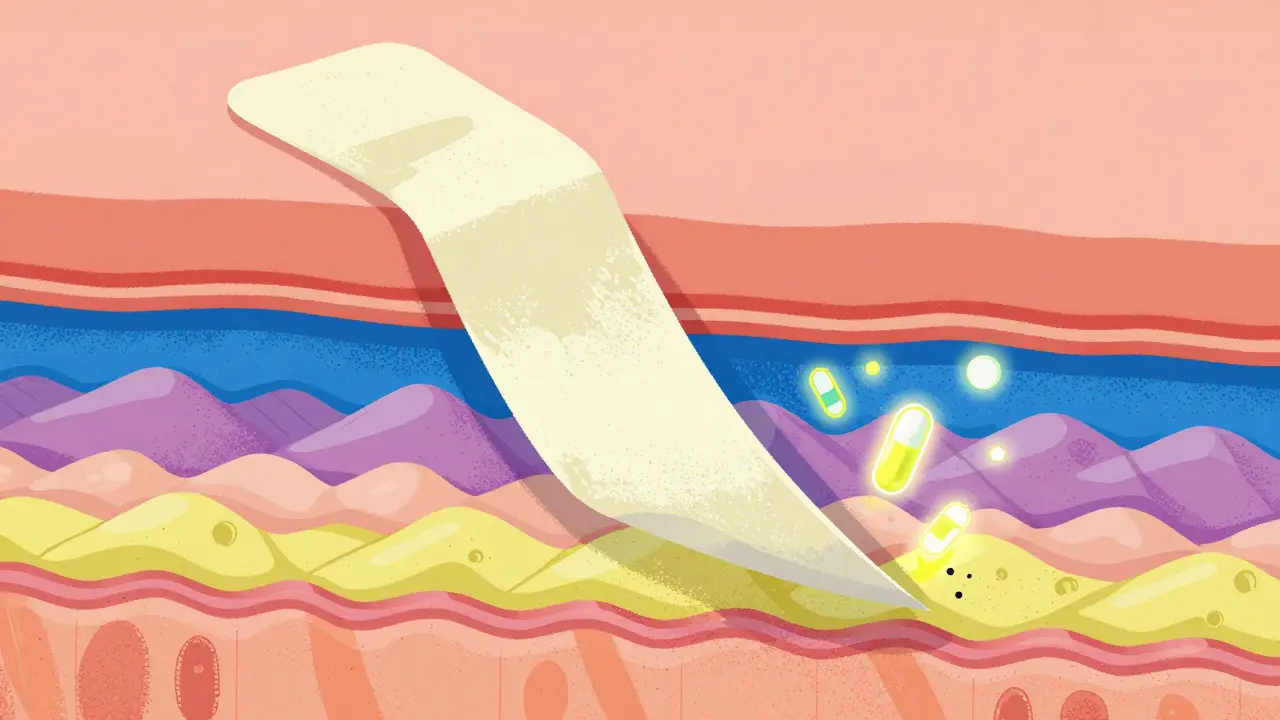
The Struggle with Topical Formulations
If you think pills are tough, try creams, ointments, or foams. For topical products, the "bloodstream" isn't the only target. You have to prove the drug actually reaches the skin layers it's intended for. Specifically, the Stratum Corneum (the outermost layer of the epidermis) is the primary barrier.
The current gold standard for testing this is tape-stripping. Basically, researchers apply adhesive tapes to the skin to peel away 15-20 sequential layers of the stratum corneum to measure drug penetration. But here's the catch: there's still a lot of gray area regarding exactly how deep you need to go or how much material to analyze. This inconsistency is why some developers see their timelines stretch by two years or more. For instance, a generic foam for psoriasis might fail three times in a row simply because the penetration measurements were inconsistent across study sites.
The cost is another barrier. While a standard oral BE study might cost $1-2 million, a comparative clinical endpoint study for a complex topical can run between $5 million and $10 million. For a mid-sized generic firm, that's a massive gamble.
Drug-Device Combinations: It's Not Just the Chemistry
When a drug is delivered via a device-like a Metered Dose Inhaler or a pre-filled syringe-the device itself becomes part of the bioequivalence equation. This is a Drug-Device Combination Product (DDCP). In these cases, the "user interface" is a critical entity. If a generic inhaler's button is slightly stiffer or the aerosol spray is a different angle, the patient might not get the full dose, regardless of how pure the drug is.
Testing focuses heavily on aerodynamic particle size distribution. If the droplets are too large, they hit the back of the throat; too small, and they are exhaled. To pass, the generic must typically maintain 80-120% of the reference product's performance metrics. Interestingly, about 65% of FDA rejection letters for DDCPs cite deficiencies in the user interface assessment, proving that the plastic and springs are just as important as the molecules.
| Feature | Single-Entity Oral | Fixed-Dose Combination (FDC) | Topical/Complex | Drug-Device (DDCP) |
|---|---|---|---|---|
| Typical Sample Size | 24-36 volunteers | 40-60 volunteers | 200-300 patients | Variable (User-focused) |
| Key Metric | Cmax / AUC | Multiple Analytes / Cmax | Skin Penetration / Endpoints | Particle Size / Device Flow |
| BE Window | 80-125% (Standard) | 80-125% per component | Often Clinical Endpoints | 80-120% (Device specs) |
| Failure Risk | Low to Moderate | High (Interactions) | Very High (Penetration) | High (UI/UX Issues) |
Navigating the Regulatory Minefield
The U.S. Food and Drug Administration (or FDA) and the European Medicines Agency (or EMA) have different appetites for risk. The EMA often requires more clinical data for complex products, which can add 15-20% to the overall development cost. If you're launching globally, you can't just do one study and hope for the best.
To survive this process, smart companies are moving toward physiologically-based pharmacokinetic (PBPK) modeling. Instead of relying solely on human trials, they use computer models to predict how a drug will behave. This has already helped reduce the number of required clinical studies by 30-50% in several approved applications. It's essentially using math to bridge the gap where human variability makes the data too noisy.
Early engagement is also key. Type II meetings with regulators have surged recently because developers realized that guessing the BE pathway is a recipe for failure. The "one-size-fits-all" approach is dead; today, you need a product-specific strategy that accounts for the specific physics and chemistry of your combination.

Common Pitfalls to Avoid
- Ignoring API Interactions: Don't assume that because Drug A and Drug B work fine alone, they will work fine together. Test for incompatibility early in the preclinical phase.
- Underestimating the Device: If you're making a DDCP, don't treat the device as a "container." It is a delivery system. Conduct rigorous human-factors engineering studies.
- Relying on Small Sample Sizes: For FDCs, a small group of volunteers might not show the variance caused by the interaction of two drugs. Over-sample to avoid a late-stage statistical failure.
- Neglecting Local Regulations: Don't assume FDA approval means a clear path at the EMA. Check for specific clinical endpoint requirements in Europe early on.
Why do combination products fail bioequivalence more often than single drugs?
It usually comes down to interactions. In Fixed-Dose Combinations, two active ingredients can interfere with each other's solubility or absorption. In drug-device products, the physical delivery mechanism adds another layer of failure; if the device doesn't deliver the drug consistently, the bioequivalence fails even if the chemical formulation is perfect.
What is the 80-125% rule in bioequivalence?
This is the standard regulatory window. It means that the 90% confidence interval for the ratio of the generic product's mean Cmax (maximum concentration) and AUC (area under the curve) must fall between 80% and 125% of the reference product's mean. For narrow therapeutic index drugs, this window is much tighter (often 90-111%).
How does PBPK modeling help in BE testing?
Physiologically-based pharmacokinetic (PBPK) modeling uses mathematical simulations of the human body (like gut pH, blood flow, and organ size) to predict drug absorption. This helps developers optimize the formulation before starting human trials and can sometimes reduce the number of clinical subjects needed by proving the mechanism of action via a model.
What is tape-stripping in topical BE studies?
Tape-stripping is a technique used to measure how much of a drug actually penetrates the skin. Researchers apply specialized adhesive tapes to remove sequential layers of the stratum corneum. These tapes are then analyzed to quantify the drug concentration at different depths, proving the generic penetrates the skin similarly to the brand-name version.
What are the most common reasons for DDCP rejection?
The most common reason is deficiencies in the comparative user interface assessment. This means the regulator found that the generic device's design, ergonomics, or mechanical performance (like the spray pattern of an inhaler) differed too much from the reference product, potentially affecting the dose the patient actually receives.
Next Steps for Developers
If you're currently in the development phase for a complex combination product, your first move should be a gap analysis. Compare your current formulation's solubility and stability against the reference product's known data. If you're developing a DDCP, start your user-interface testing immediately-don't wait until the drug is finalized.
For those facing repeated BE failures, consider pivoting to an in vitro-in vivo correlation (IVIVC) approach. While not always accepted, showing that your lab results (in vitro) strongly predict human results (in vivo) can give you the insight needed to tweak the formulation without spending another $5 million on a failed human trial.
13 Comments
Mike ArrantApril 23, 2026 AT 02:15
Most of these companies just throw money at the wall to see what sticks. If you can't get the device UI right, you're basically just playing house with chemistry. Total amateur hour.
Chidi ProsperApril 25, 2026 AT 00:58
The PBPK modeling mentioned is absolutely the way forward. It cuts down the waste and gets us to the goal faster. We need more of this tech-driven approach in the industry.
Rick BrewsterApril 26, 2026 AT 04:10
one must consider that the mere act of measuring a thing is to change the thing itself and thus the tape stripping process is but a dance of shadows where the truth of the stratum corneum remains elusive because the observer is always an interloper in the biological sanctuary of the skin and honestly the fda just wants to maintain a hegemony of bureaucratic stasis thru endless iterations of pointless data
Mayur Pankhi SaikiaApril 26, 2026 AT 06:20
Actually... PBPK modeling is overated!! It's just a way for lazy developers to avoid real human data... just a fancy spreadsheet that hides the real variability!!
Anantha LakshmiApril 27, 2026 AT 20:13
Let's stay positive about these hurdles! 🌟 Every failure is just a step closer to a better product for the patients. We can do this! 💪✨
Dave EdwardsApril 28, 2026 AT 00:57
Imagine spending 10 million on a cream and still failing. Absolute madness. Why are we even allowing this level of waste in the first place? 🙄
Divyanshu GiriApril 28, 2026 AT 10:38
Keep pushing the boundaries folks! This stuff is tough but the payoff for patients is huge! Just keep grinding and the wins will come!
Sarah WattersApril 30, 2026 AT 04:45
Funny how the FDA and EMA just happen to disagree on everything. Probably just a way to keep the big pharma lobbyists in control of the global supply chain so they can hike prices in Europe.
Saptatshi BiswasMay 1, 2026 AT 20:36
The sheer incompetence of firms failing the user interface tests is staggering. It is a basic engineering requirement, yet they treat it as an afterthought. Truly pathetic.
Mel GlickMay 2, 2026 AT 00:31
The 80-125% window is an industry standard for a reason. Trying to bypass it or complain about it is just ignoring the basic science of pharmacology. Deal with it.
Sue StollerMay 2, 2026 AT 12:35
It's so inspiring to see the progress in modeling! 🌈 It'll make things so much more accessible for smaller firms too. Keep dreaming big! ✨😊
Nicole AntunesMay 3, 2026 AT 07:55
The focus on human-factors engineering is a very positive step toward patient safety. It ensures that the medication is not only chemically sound but also practically usable. :)
Amy FredericksMay 4, 2026 AT 17:43
I totally agree with the point about human-factors engineering. It really is about making sure the person at home can actually use the device without stress. When we prioritize the patient's experience, the regulatory hurdles start to feel more like a safety net than a wall. It's all about that inclusive design process where we consider every possible user, regardless of their dexterity or age. If a senior citizen can't press that button, the most pure drug in the world is useless. We should be mentoring new developers to think about the human element from day one. Let's keep supporting each other through these complex trials. It takes a village to get a safe generic to market, and seeing the community share these guides is just wonderful. We're all in this together to make healthcare better. Every little bit of knowledge shared here helps someone avoid a costly mistake. It's just so heartening to see the transparency in these discussions. Let's keep the energy high and the spirits higher as we navigate these FDA minefields together. We've got this!